Comprender los nódulos de Lisch: síntomas, causas y tratamientos para el diagnóstico de neurofibromatosis
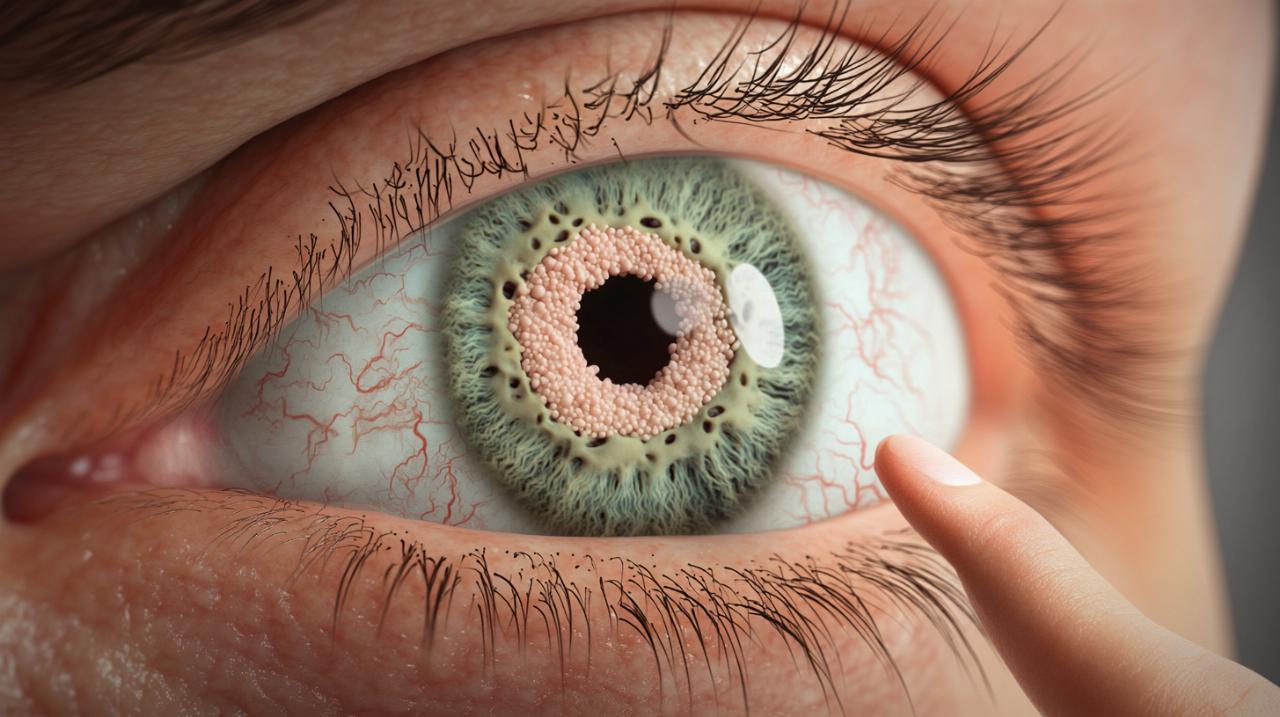
Los nódulos de Lisch representan una característica oftalmológica que ha ganado relevancia en el campo de la medicina por su estrecha asociación con una condición genética que afecta a miles de personas en todo el mundo. Estas formaciones benignas en el iris no solo son un signo clínico de gran valor diagnóstico, sino que también abren una ventana para comprender mejor el complejo espectro de las enfermedades relacionadas con mutaciones genéticas. Conocer su naturaleza, cómo se manifiestan y qué papel desempeñan en el diagnóstico de ciertas condiciones es fundamental para pacientes, familias y profesionales de la salud que buscan una atención integral y personalizada.
¿Qué son los nódulos de Lisch y cómo se relacionan con la neurofibromatosis?
Definición y características de los nódulos de Lisch en el iris
Los nódulos de Lisch son hamartomas benignos que se desarrollan en el iris, la parte coloreada del ojo. Estas formaciones se presentan como manchas marrones o amarillentas que pueden ser observadas durante un examen oftalmológico especializado. A diferencia de otras lesiones oculares, los nódulos de Lisch no comprometen la función visual y no representan un riesgo directo para la salud ocular del paciente. Sin embargo, su presencia constituye un marcador clínico altamente específico que puede orientar hacia el diagnóstico de una enfermedad genética subyacente. La textura y el patrón de estas lesiones son tan distintivos que su detección durante la exploración oftalmológica ofrece información valiosa para el médico especialista.
Durante la adolescencia y la vida adulta es cuando estos nódulos comienzan a aparecer de manera más evidente, facilitando su identificación mediante el uso de tecnología oftalmológica avanzada. Aunque no causan síntomas directos, su detección temprana permite iniciar un seguimiento adecuado y establecer estrategias de manejo preventivo para otras manifestaciones asociadas a la condición genética que representan. Los profesionales de la salud suelen utilizar la lámpara de hendidadura para visualizar con precisión estas estructuras en el iris, lo que garantiza un diagnóstico certero y oportuno.
Relación entre los nódulos de Lisch y la neurofibromatosis tipo 1
La neurofibromatosis tipo 1, conocida también como NF1, es una enfermedad autosómica dominante que se manifiesta con una variedad de signos clínicos, entre los cuales los nódulos de Lisch ocupan un lugar destacado. Esta condición genética es causada por mutaciones en el gen NF1, localizado en el cromosoma 17, el cual codifica una proteína denominada neurofibromina. Esta proteína cumple funciones importantes en la regulación del crecimiento celular, y cuando su producción se ve afectada, se desencadenan múltiples manifestaciones clínicas que pueden comprometer diferentes sistemas del organismo.
La presencia de nódulos de Lisch en el iris se considera uno de los criterios diagnósticos establecidos por el Instituto Nacional de la Salud de Estados Unidos para confirmar la neurofibromatosis tipo 1. Estos nódulos forman parte de un conjunto de signos clínicos que incluyen también manchas café con leche, neurofibromas dérmicos y neurofibromas plexiformes, así como efélides axilares conocidas como signo de Crowe. La detección de dos o más nódulos de Lisch durante el examen oftalmológico, junto con la presencia de otros criterios clínicos, permite al médico establecer un diagnóstico con alta confianza. Es importante destacar que estos nódulos aparecen de forma progresiva durante la adolescencia y en la vida adulta, lo que refuerza la necesidad de realizar evaluaciones oftalmológicas periódicas en pacientes con antecedentes familiares de NF1 o con otros signos sugestivos de la enfermedad.
Síntomas y signos clínicos de los nódulos de Lisch
Manifestaciones visuales y métodos de detección oftalmológica
Una de las particularidades más notables de los nódulos de Lisch es que, en términos generales, no afectan la visión ni producen molestias perceptibles para el paciente. Esta ausencia de síntomas directos puede retrasar su detección si no se realizan exámenes oftalmológicos de rutina o dirigidos. La identificación de estos nódulos requiere de una exploración detallada del iris mediante el uso de la lámpara de hendidura, un instrumento que permite a los oftalmólogos visualizar con gran precisión las estructuras oculares. Durante este examen, el especialista busca la presencia de lesiones pigmentadas que presenten las características morfológicas típicas de los hamartomas benignos asociados a la neurofibromatosis tipo 1.
El procedimiento de evaluación oftalmológica es indoloro y no invasivo, lo que facilita su realización incluso en pacientes pediátricos. En muchos casos, la detección de los nódulos de Lisch ocurre de manera incidental durante revisiones oftalmológicas de rutina, especialmente cuando existe un historial familiar conocido de neurofibromatosis tipo 1. La evaluación debe ser realizada por un oftalmólogo con experiencia en el diagnóstico de condiciones genéticas que afectan el sistema visual, ya que la interpretación correcta de los hallazgos clínicos es fundamental para establecer un diagnóstico preciso y orientar el manejo multidisciplinario del paciente.
Diferencias entre los nódulos de Lisch y otras lesiones del iris
El iris puede presentar diversas alteraciones pigmentarias que en ocasiones pueden generar confusión en el diagnóstico diferencial. Sin embargo, los nódulos de Lisch poseen características muy específicas que permiten diferenciarlos de otras lesiones benignas o malignas que pueden aparecer en esta estructura ocular. A diferencia de los nevos del iris, que suelen presentar un patrón de pigmentación más uniforme y pueden aparecer en ausencia de condiciones genéticas subyacentes, los nódulos de Lisch tienen un aspecto más irregular y tienden a aumentar en número con el paso del tiempo en pacientes con neurofibromatosis tipo 1.
Otra diferencia relevante radica en la asociación clínica de los nódulos de Lisch con otros signos cutáneos y sistémicos propios de la NF1. La presencia de múltiples manchas café con leche, neurofibromas cutáneos y efélides en áreas como las axilas o la ingle refuerza la sospecha diagnóstica de que las lesiones observadas en el iris corresponden efectivamente a nódulos de Lisch y no a otras alteraciones pigmentarias aisladas. El contexto clínico global del paciente, incluyendo su historial médico personal y familiar, juega un papel crucial en la interpretación de los hallazgos oftalmológicos y en la formulación de un plan diagnóstico y terapéutico adecuado.
Causas y factores de riesgo asociados a los nódulos de Lisch
Base genética de la neurofibromatosis tipo 1 y desarrollo de nódulos
La aparición de nódulos de Lisch en el iris está directamente relacionada con mutaciones en el gen NF1, localizado en el cromosoma 17 en la posición q11.2. Este gen es responsable de codificar la neurofibromina, una proteína que actúa como un regulador negativo de la proliferación celular al inhibir la actividad de la proteína RAS. Cuando se producen mutaciones en el gen NF1, la neurofibromina pierde su función reguladora, lo que resulta en un crecimiento celular descontrolado que se manifiesta a través de los múltiples signos clínicos de la neurofibromatosis tipo 1, incluyendo el desarrollo de nódulos de Lisch en el iris.
La neurofibromatosis tipo 1 se hereda de forma autosómica dominante, lo que significa que basta con que uno de los progenitores transmita la mutación para que el descendiente tenga una alta probabilidad de desarrollar la enfermedad. Hasta la fecha se han identificado más de doscientas cuarenta mutaciones distintas en el gen NF1, lo que explica la gran variabilidad en la presentación clínica de la enfermedad entre diferentes individuos, incluso dentro de la misma familia. A pesar de que el diagnóstico genético puede detectar alteraciones en el gen NF1 con una precisión cercana al noventa por ciento, no es posible predecir con exactitud el fenotipo que desarrollará cada paciente, es decir, la gravedad y el tipo de manifestaciones clínicas que presentará a lo largo de su vida.
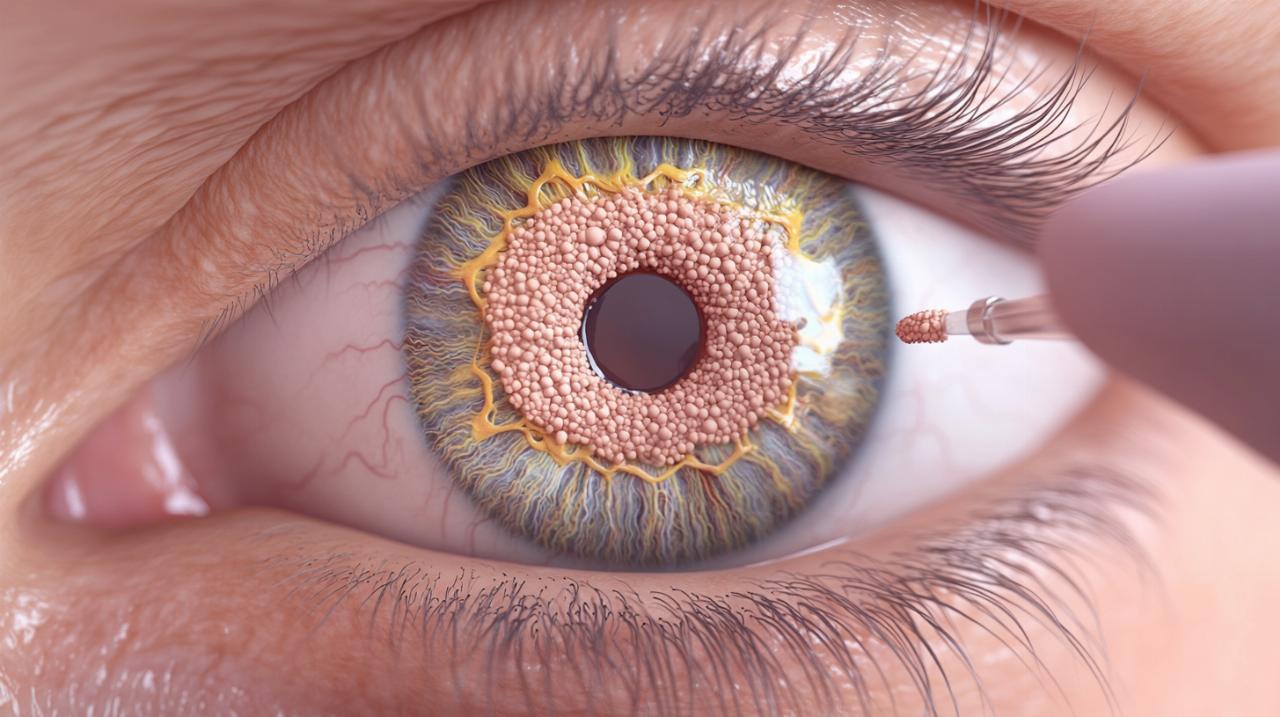
Edad de aparición y prevalencia en pacientes con neurofibromatosis
Los nódulos de Lisch no están presentes desde el nacimiento, sino que comienzan a manifestarse durante la adolescencia y continúan acumulándose en la vida adulta. Esta característica temporal tiene importantes implicaciones para el diagnóstico y seguimiento de los pacientes con sospecha de neurofibromatosis tipo 1. En la infancia temprana, es posible que estos nódulos no sean visibles, por lo que los criterios diagnósticos se basan principalmente en la detección de otros signos clínicos como las manchas café con leche o los neurofibromas cutáneos. Sin embargo, a medida que el paciente madura, la aparición de los nódulos de Lisch se convierte en un criterio diagnóstico cada vez más relevante.
La neurofibromatosis tipo 1 afecta aproximadamente a una de cada tres mil personas, lo que la convierte en una de las enfermedades genéticas más frecuentes con impacto significativo en múltiples sistemas del organismo. En pacientes con diagnóstico confirmado de NF1, la prevalencia de nódulos de Lisch es muy elevada, especialmente en la edad adulta, donde prácticamente todos los pacientes desarrollan al menos algunos de estos hamartomas benignos en el iris. Esta alta prevalencia refuerza la importancia de realizar evaluaciones oftalmológicas periódicas en todos los individuos con antecedentes personales o familiares de neurofibromatosis tipo 1, ya que la detección temprana de los nódulos de Lisch puede facilitar el diagnóstico y permitir el inicio de un seguimiento médico integral.
Diagnóstico y opciones de tratamiento para los nódulos de Lisch
Procedimientos de examen oftalmológico y criterios diagnósticos
El diagnóstico de los nódulos de Lisch se realiza mediante un examen oftalmológico detallado utilizando la lámpara de hendidura, un instrumento que permite la visualización de las estructuras del ojo con gran precisión. Durante la exploración, el oftalmólogo busca la presencia de lesiones pigmentadas características en el iris, que se manifiestan como manchas de color marrón o amarillento. La identificación de dos o más nódulos de Lisch en un paciente es altamente sugestiva de neurofibromatosis tipo 1, especialmente cuando se acompaña de otros signos clínicos establecidos en los criterios diagnósticos del Instituto Nacional de la Salud de Estados Unidos.
Estos criterios diagnósticos incluyen la presencia de seis o más manchas café con leche con un diámetro superior a cinco milímetros en niños o superior a quince milímetros en adolescentes y adultos, la existencia de dos o más neurofibromas de cualquier tipo o al menos un neurofibroma plexiforme, la presencia de efélides en áreas como axilas o ingles, la detección de un glioma óptico, la identificación de dos o más nódulos de Lisch, la presencia de anomalías óseas características y la existencia de un familiar de primer grado con diagnóstico confirmado de NF1. El cumplimiento de dos o más de estos criterios permite establecer el diagnóstico con un alto grado de certeza, siendo los criterios útiles en aproximadamente el noventa y cuatro por ciento de los casos para la edad de seis años.
En algunos casos, cuando la presentación clínica no es clara o cuando existe la necesidad de confirmar el diagnóstico para asesoramiento genético, se pueden realizar pruebas genéticas que buscan mutaciones en el gen NF1. Estas pruebas pueden detectar alteraciones genéticas con una efectividad de hasta el noventa por ciento, aunque no permiten predecir la severidad de las manifestaciones clínicas que desarrollará el paciente. El diagnóstico temprano es fundamental para implementar un plan de seguimiento adecuado que incluya evaluaciones oftalmológicas, neurológicas, dermatológicas y ortopédicas, con el fin de detectar y manejar de manera oportuna cualquier complicación que pueda surgir.
Seguimiento médico y manejo terapéutico de pacientes con nódulos de Lisch
Los nódulos de Lisch en sí mismos no requieren tratamiento específico, ya que son hamartomas benignos que no afectan la visión ni producen complicaciones oculares directas. Sin embargo, su presencia indica la existencia de neurofibromatosis tipo 1, una condición que requiere un seguimiento médico continuo y multidisciplinario para prevenir y manejar las múltiples complicaciones que pueden surgir a lo largo de la vida del paciente. El manejo de la NF1 incluye evaluaciones oftalmológicas periódicas para detectar posibles gliomas ópticos, controles dermatológicos para vigilar el desarrollo de neurofibromas cutáneos, evaluaciones neurológicas para identificar dificultades de aprendizaje o convulsiones, y revisiones ortopédicas para detectar anomalías esqueléticas.
En niños con neurofibromatosis tipo 1 se recomiendan controles médicos anuales que incluyan revisión de la piel, medición de la presión arterial, evaluación del crecimiento y desarrollo, detección de signos de pubertad precoz, análisis de posibles cambios en el esqueleto y exámenes oculares completos. Estas evaluaciones permiten identificar de manera temprana cualquier complicación y aplicar las intervenciones terapéuticas necesarias. En el caso de neurofibromas plexiformes que causan síntomas significativos o comprometen funciones vitales, se puede considerar el tratamiento con selumetinib, un medicamento aprobado por la Administración de Alimentos y Medicamentos de Estados Unidos para pacientes pediátricos a partir de los dos años de edad con NF1 y neurofibromas plexiformes inoperables.
Cuando los tumores benignos o malignos asociados a la NF1 causan complicaciones graves, la cirugía puede ser necesaria para extirparlos y aliviar los síntomas. Los tumores malignos de la vaina del nervio periférico, que afectan aproximadamente al diez por ciento de las personas con NF1, requieren tratamiento oncológico especializado que puede incluir cirugía, radioterapia y quimioterapia. En los últimos años se han realizado avances significativos en la investigación de terapias génicas y tratamientos dirigidos que podrían ofrecer nuevas alternativas terapéuticas en el futuro. El pronóstico de los pacientes con neurofibromatosis tipo 1 varía considerablemente en función de la gravedad de las manifestaciones clínicas y de la detección temprana de las complicaciones, pero con un seguimiento médico adecuado y una atención multidisciplinaria es posible mejorar significativamente la calidad de vida y la supervivencia de quienes viven con esta condición genética.